


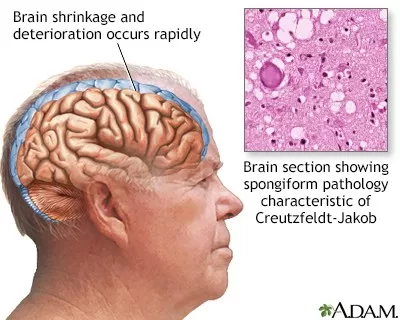
Creutzfeldt-Jakob Disease (CJD) is a rare but devastating neurological disorder characterized by rapid cognitive decline and severe neurological impairment. Often classified among prion diseases, CJD symptoms can initially mimic other conditions, making diagnosis a complex process. Understanding CJD diagnosis and prognosis is crucial for affected individuals and their families, particularly given the bleak outlook following diagnosis. The average survival time post-diagnosis ranges from a few months to two years, emphasizing the urgency for research and awareness of this condition. In this article, we will delve deeper into the symptoms, diagnostic strategies, and the current state of CJD management, aiming to shed light on this enigmatic disease.
Creutzfeldt-Jakob Disease, also known as CJD, manifests as one of the most challenging prion diseases to diagnose due to its nonspecific symptoms. Affiliated with various neurological disorders, CJD can be sporadic, familial, or acquired through exposure to infected tissue. The complexities of prion disease symptoms often lead to misdiagnoses, emphasizing the importance of precise evaluation during the diagnostic process. Families dealing with familial CJD may face unique challenges associated with genetic testing and hereditary risks. In this discussion, we will explore the intricacies surrounding CJD, its impact on neurological health, and the promising avenues of ongoing research aimed at enhancing our understanding of this fatal condition.
Understanding Creutzfeldt-Jakob Disease Symptoms
Creutzfeldt-Jakob Disease (CJD) symptoms are often misinterpreted as part of the normal aging process, which complicates dedicated and accurate diagnosis. Early stages may present with subtle cognitive impairments and personality changes that are easily mistaken for stress or depression. As the disease progresses, distinctive neurological signs such as myoclonus and visual disturbances become more apparent. Understanding these symptoms is key for both healthcare professionals and caregivers, as recognizing them can lead to timely medical attention and potential participation in clinical trials.
Patients suffering from CJD may also experience severe memory loss and unpredictable behavior, leading to significant challenges for families. Communication can become more difficult as the patient’s ability to express themselves declines. Moreover, neurological decline can manifest physically through stiffness and coordination issues, contributing to a reduced quality of life. Awareness of these symptoms not only aids in diagnosis but also enhances support for those affected and their loved ones.
Frequently Asked Questions
What are the symptoms of Creutzfeldt-Jakob Disease (CJD)?
Creutzfeldt-Jakob Disease (CJD) is characterized by rapidly progressing symptoms including cognitive decline, personality changes, memory loss, severe neurological dysfunction, visual disturbances, muscle stiffness, and involuntary movements. These prion disease symptoms typically lead to significant deterioration in quality of life.
How is CJD diagnosed and what tests are used?
Diagnosing Creutzfeldt-Jakob Disease (CJD) involves a combination of neurological exams, MRI scans, EEG, and cerebrospinal fluid (CSF) analysis. MRI typically reveals characteristic brain changes, while CSF testing can detect abnormal proteins associated with prion diseases. Together, these diagnostic methods help differentiate CJD from other neurological disorders.
What is the prognosis for someone diagnosed with CJD?
The prognosis for Creutzfeldt-Jakob Disease (CJD) is generally poor, with an average survival time after diagnosis ranging from a few months to two years. This rapid progression can vary depending on the specific variant of CJD and individual patient factors. Sporadic CJD typically has a swift onset and progression.
Is there any treatment available for Creutzfeldt-Jakob Disease?
Currently, there are no effective treatments to cure or significantly slow down Creutzfeldt-Jakob Disease (CJD). Management is mainly focused on palliative care to alleviate symptoms and improve the quality of life for patients and their families. Research is ongoing to identify potential neuroprotective agents and other therapeutic options.
What causes familial Creutzfeldt-Jakob Disease?
Familial Creutzfeldt-Jakob Disease (CJD) is caused by genetic mutations in the PRNP gene, which can be inherited within families. Individuals with a family history of this prion disease are more likely to undergo genetic testing to assess their risk for developing CJD and to receive genetic counseling.
| Key Points | |
|---|---|
| Diagnosis Challenges | Determining CJD can be difficult due to similar symptoms with other neurological disorders. |
| Diagnostic Tests | Includes neurological exams, MRI scans, EEG, and CSF analysis. |
| Genetic Testing | Identifies familial CJD risks through PRNP gene mutations. |
| Prognosis Overview | Average survival ranges from months to two years after diagnosis. |
| Progression Symptoms | Initial symptoms include cognitive decline, leading to severe neurological issues. |
| Current Treatment Options | No known cure; focus on palliative care to improve quality of life. |
| Research and Awareness | Continued research is needed for better diagnostics and treatment. |
Summary
Creutzfeldt-Jakob Disease (CJD) is a rare neurological disorder that poses significant diagnostic and prognostic challenges due to its swift progression and complexity. Understanding CJD is vital, as it helps illuminate the symptoms, diagnostic methods, and the current lack of effective treatments. Efforts to increase public awareness and research can lead to better understanding and, ultimately, improved outcomes for those affected by this devastating disease.
The content provided on this blog (e.g., symptom descriptions, health tips, or general advice) is for informational purposes only and is not a substitute for professional medical advice, diagnosis, or treatment. Always seek the guidance of your physician or other qualified healthcare provider with any questions you may have regarding a medical condition. Never disregard professional medical advice or delay seeking it because of something you have read on this website. If you believe you may have a medical emergency, call your doctor or emergency services immediately. Reliance on any information provided by this blog is solely at your own risk.